
近日,中国科学院上海药物研究所研究员陈勋团队联合日本京都大学、加拿大麦吉尔大学科研人员,通过开发基于进化的转座子注释新方法,并结合系统发育分析、大规模平行报告系统及多组学等技术,在单碱基水平上揭示了内源性逆转录病毒序列和功能在不同物种间的差异进化。
人体内病毒组包括感染性病毒、整合到人基因组中的外源病毒以及内源性逆转录病毒等多种类型。内源性逆转录病毒序列是转座子重复序列的一种,占据了人基因组的8%,其来源于百万年前感染人类祖先的逆转录病毒,因在漫长的进化过程中被“捕获”而保留在基因组中。多种古老病毒能够感染并被“捕获”,其中包括在不同进化阶段感染人类祖先的HERV-E、HERV-K及HERV-H等多种类型。根据序列差异,这些内源性逆转录病毒序列可被分为500多个亚家族。
通常,完整的内源性逆转录病毒序列包括gag、pol及env三个核心基因结构域,以及两端具有重要调控功能的长末端重复序列(LTRs)。大多数内源性逆转录病毒序列在进化过程中,因突变、重组等失去原有功能。因此,内源性逆转录病毒序列曾被学界认为是基因组中无功能的“垃圾”序列。然而,大量研究表明,内源性逆转录病毒尤其是LTR区域富集了众多转录因子结合位点,可作为顺式作用元件调控邻近基因表达,并参与先天免疫等多个基因表达调控网络与生物学过程。因此,内源性逆转录病毒在癌症、发育及感染性疾病等多种人类疾病中发挥重要作用,准确注释内源性逆转录病毒序列是其功能与进化研究的基础。但目前,主要依赖序列比对的注释方法存在局限性,使人基因组内源性逆转录病毒序列数据库中存在大量注释错误。
为解决上述问题,研究人员开发了转座子序列注释新方法。该方法利用系统发育分析,将序列相近但被误分至不同亚家族的序列进行整合和重新注释。研究人员针对76个进化上较年轻的内源性逆转录病毒亚家族,成功应用该方法纠正了其中26个亚家族近三分之一的序列注释错误。同时,研究以MER11家族为例,结合系统发育分析与表观基因组学数据,揭示了MER11A/B/C亚家族中大量注释错误,并利用新方法进行了校正。研究人员鉴定出四个新的亚家族:MER11_G1、G2、G3、G4。基于这些更准确的注释信息,研究人员能够对MER11家族表观遗传修饰特征进行更精细的分组研究并追溯其进化历史。
进一步,研究人员利用大规模平行报告系统技术实验验证了来自人、大猩猩及猕猴基因组中共7000多条内源性逆转录病毒MER11序列的转录调控活性,并在单碱基分辨率水平上揭示了与之相关的SOXs等转录因子结合位点序列及其进化规律。研究显示,研究人员在进化上较年轻的MER11序列中鉴定出人类与大猩猩特有的SOXs结合位点序列。该SOXs结合位点序列由灵长类物种进化过程中单碱基缺失事件产生,并显著增强了其调控活性。
该研究建立了内源性逆转录病毒等转座子分类、注释、进化以及生物学功能研究的新方法。利用这一方法,科研人员能够更准确地追踪同一内源性逆转录病毒序列及其功能演化历史,且获得的精准的内源性逆转录病毒注释信息将显著提升后续序列和功能鉴定可靠性,有助于学界系统性研究内源性逆转录病毒在肿瘤发生、发育调控、免疫相关疾病中的生物学作用及其进化。同时,该方法也可被用于深入研究流感等病原体的突变模式、演化动态及功能特征。下一步,研究团队将结合系统发育学、多组学及人工智能等前沿技术,全面解析内源性逆转录病毒的复杂生物学功能,探讨其中的隐藏靶点,并研究其在人类免疫系统中的重要作用。
相关研究成果以A phylogenetic approach uncovers cryptic endogenous retrovirus subfamilies in the primate lineage为题,发表在《科学进展》(Science Advances)上。
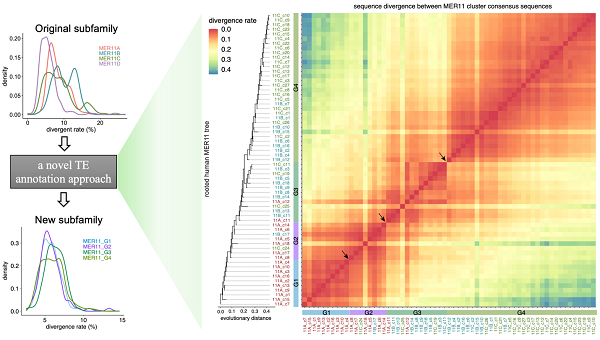
基于进化的转座子序列注释新方法
供稿人:杨越
审核人:文成锋


